

《除菌過濾技術及應用指南》之除菌過濾驗證
為指導和規范除菌過濾技術在無菌藥品生產中的應用,保證無菌藥品的安全、有效和質量穩定,依據《藥品生產質量管理規范(2010年修訂)》及附錄,制定本指南。本指南中的除菌過濾是指采用物理截留的方法去除液體或氣體中的微生物,以達到無菌藥品相關質量要求的過程。包括除菌過濾系統的設計、選擇、驗證、使用等內容,適用于無菌藥品從工藝開發到上市生產的整個生命周期。
相關除菌過濾器產品
|
|
|
|
PTFE除菌濾芯 |
除菌級(PES)囊式濾芯 |


除菌過濾器—除菌過濾驗證概述
|
概述 |
圖片 |
注意事項 |
|
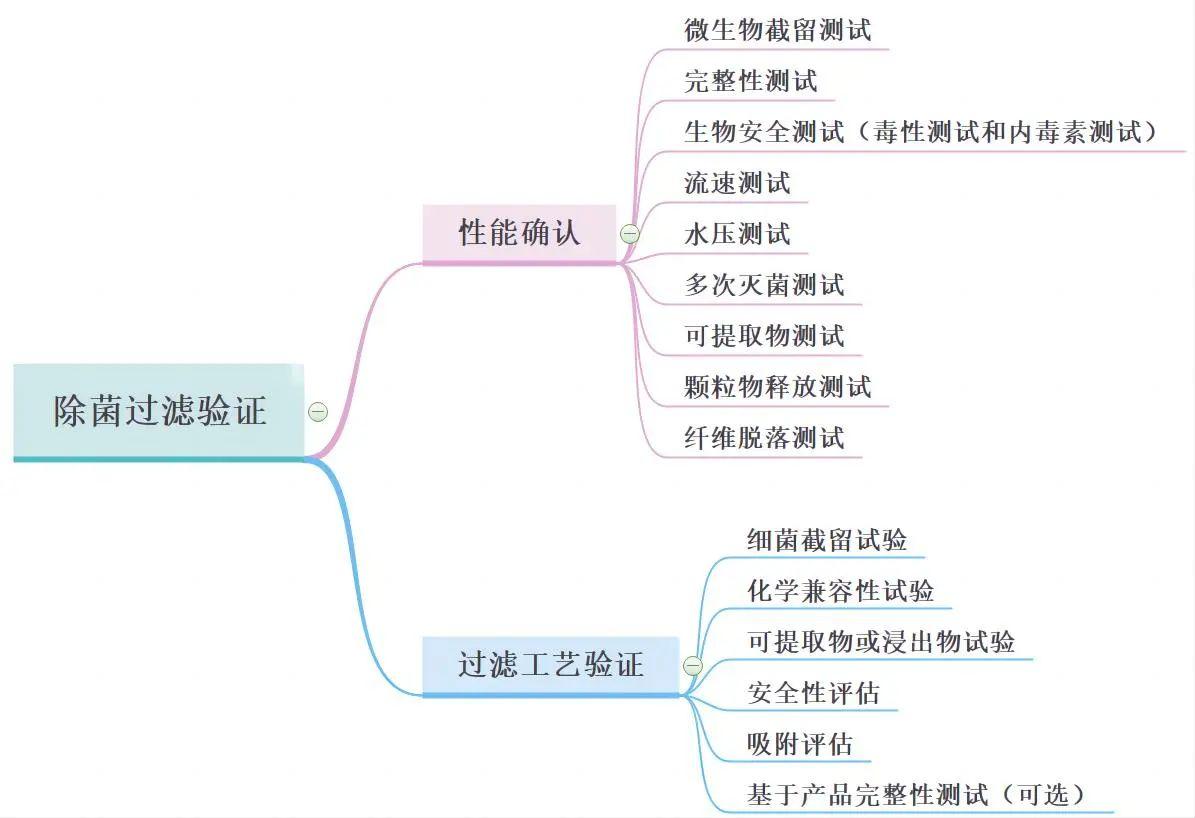
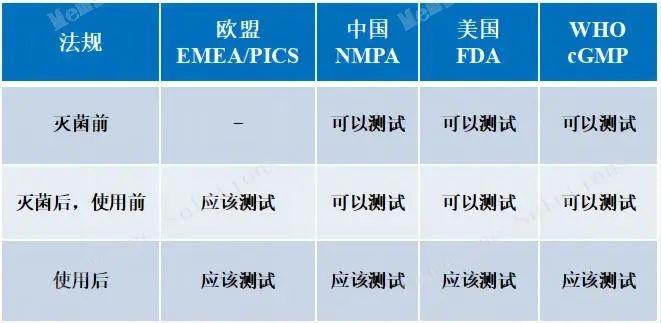
除菌過濾驗證包含以下2類,由于除菌過濾器性能確認和過濾工藝驗證,兩者很難互相替代,因此應獨立完成。
2、過濾工藝驗證:除菌過濾工藝驗證可以由過濾器的使用者或委托試驗檢測機構(例如:過濾器的生產者或第三方試驗室)完成,但過濾器使用者應最終保證實際生產過程中操作參數和允許的極值在驗證時已被覆蓋,并有相應證明文件。 |
|
不同過濾器生產商的驗證文件一般是不能相互替代的,同一生產商的同一材質的除菌過濾驗證文件往往也不能直接互換,除非有合理的聲明或文件支持。
|
除菌過濾器—細菌截留試驗
|
目的 |
圖片 |
注意事項 |
|
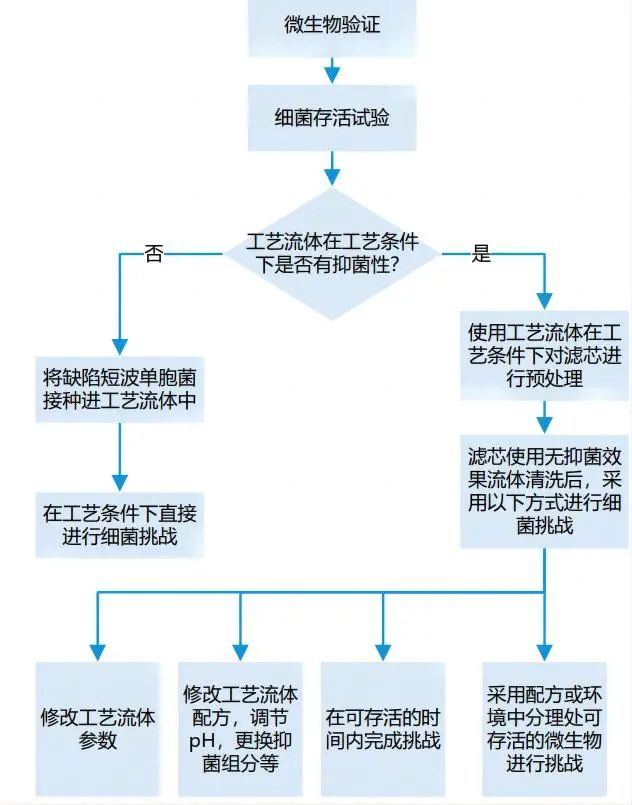
模擬實際生產過濾工藝中的最差條件(解讀:過濾溫度相對越高,過濾時間越長,批量越大,壓差越高越是最差條件),過濾含有一定量挑戰微生物的產品溶液或者產品替代溶液,以確認除菌過濾器的微生物截留能力。
|
|
1、應先獲得最差條件下的可提取物數據,將其用于藥品的安全性評估。可提取物反映了浸出物的最大可能,無論是否要做浸出物試驗,可提取物的測試和評估都非常重要。(解讀:可提取物是一定要做的,浸出物可以根據可提取物的測試和評估其風險,如果當風險不可忽略時,浸出物的測試也是不可避免的。) 2、用于測試的模型溶劑應能夠模擬實際的藥品處方,同時與過濾器不應有化學兼容性方面的問題。通常應具有與產品相同或相似的理化性質,如pH值、極性或離子強度等。(解讀:根據配方中的理化性質(pH,有機溶質/溶劑)和占比選擇模型溶劑,通常所選模型溶劑提取能力比實際配方更強。) 3、使用最長過濾時間、最高過濾溫度、最多次蒸汽滅菌循環、增加伽瑪輻射的次數和劑量都可能會增加可提取物水平。(解讀:可提取物的檢測影響因素比較多,平行對比測試的條件需要保持一致。) 4、可提取物試驗應使用滅菌后的濾器來完成。用于試驗的過濾器盡量不進行預沖洗。 5、可提取物和浸出物的檢測需要采用定性和定量結合的方法。 6、在完成可提取物或者浸出物試驗后,應針對過濾器可提取物或浸出物的種類和含量,結合藥品最終劑型中的濃度、劑量大小、給藥時間、給藥途徑等對結果進行安全性評估,以評估可提取物和浸出物是否存在安全性風險。 |
除菌過濾器—化學兼容性試驗
|
化學兼容性試驗的研究目的 |
圖片 |
注意事項 |
|
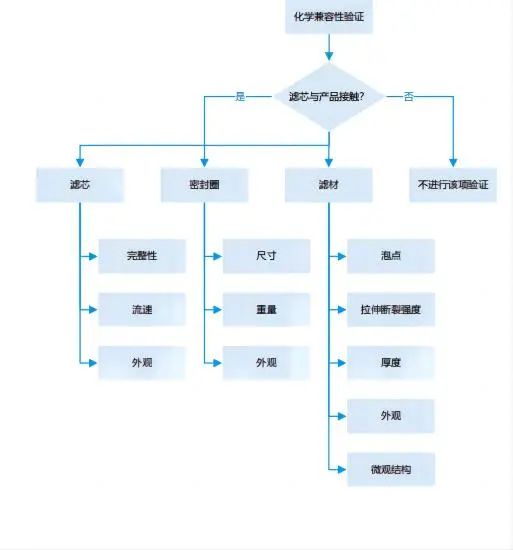
用來評估在特定工藝條件下,待過濾介質對過濾裝置的化學影響 |
|
1、化學兼容性試驗應涵蓋整個過濾裝置,不只是濾膜。(解讀:除了濾膜,其外殼骨架、密封圈、支撐導流層均需要考慮。) 3、化學兼容性試驗檢測項目一般包括:過濾器接觸待過濾介質前后的目視檢查;過濾過程中流速變化;濾膜重量/厚度的變化;過濾前后起泡點等完整性測試數值的變化;濾膜拉伸強度的變化;濾膜電鏡掃描確認等。 |
除菌過濾器—吸附試驗
|
描述 |
圖片 |
|
1、待過濾介質中的某些成分粘附在濾器上的過程,可能影響待過濾介質的組成和濃度。(解讀:吸附實驗的目的就是考察濾膜或濾芯是否會帶走藥液中的關鍵成分,從而影響最終產品的質量。) |
|
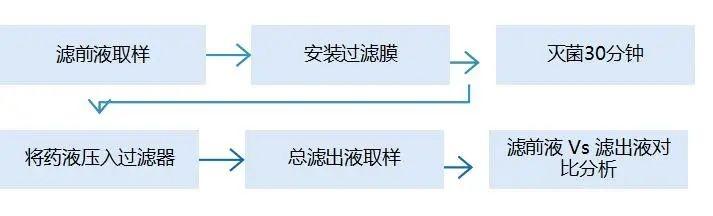
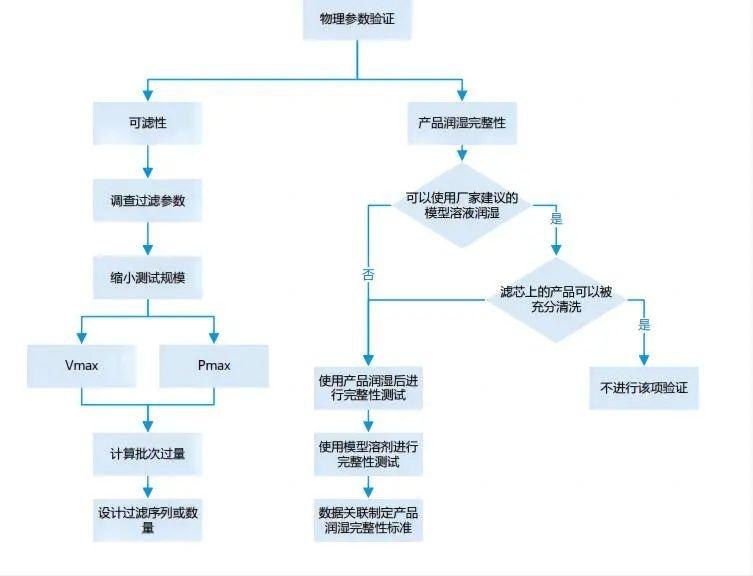
除菌過濾器—基于產品完整性試驗
|
概述 |
流程 |
|
|
除菌過濾驗證—再驗證
|
完成過濾工藝的驗證之后,還應當定期評估產品性質和工藝條件,以確定是否需要進行再驗證。 |
至少(但不限于)對以下內容進行評估,以決定是否需要開展再驗證: |
參考文獻:
[1] 《藥品生產質量管理規范》(2010年修訂)及附錄
[2] 國家藥品監督管理局《除菌過濾技術及應用指南》(2018.10)
[3] PDA Technical Report No.40,Sterilizing Filtration of Gases.
[4] PDA Technical Report No.26 Revised 2008,Sterilizing Filtration of Liquids.
[5] USFDA Guidance for Industry,Sterile Drug Products Produced by Aseptic Processing-Current Good Manufacturing Practice,2004.
[6] EU Guidelines to Good Manufacturing Practice,Annex 1 Manufacture of Sterile Medicinal Products,2008


.png)


.png)
.png)

(1).jpg)
